

Tell us about yourself




Not sure?
We can help you build your routine
Take our skin quiz
Not sure?
We can help you build your routine
 Play Video
Play Video
“I’m living life and my eczema isn’t stopping me from what I love doing.”
Living with eczema can be tough—but it doesn’t have to stop you from thriving.
At Gladskin, we’re committed to helping you build stronger, healthier skin from the microbiome up.
Big eczema hope starts on a microscopic level—with Gladskin.
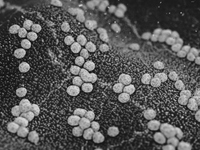
Our Science
When the bacteria on your skin gets out of balance, it can cause an eczema flare-up. Gladskin’s patented protein Micreobalance® restores bacterial balance to the skin microbiome, restoring balance to support skin’s natural healing process.
Learn more Play Video
Play Video
“I’ve been able to put Gladskin to use in my practice with great success.”
 Play Video
Play Video
A healthy skin microbiome is key to reducing flares. Gladskin’s patented endolysin science can help.

A New Kind of Eczema Cream
You’ve probably tried dozens of eczema creams to help with itchy skin and sleepless nights—and they didn’t deliver results. Gladskin Eczema Cream is clinically proven to reduce eczema symptoms. Discover eczema symptom relief from the microbiome up.
Shop Eczema
Soothing Redness-Prone Skin
Even though facial redness is common, it’s often misdiagnosed and misunderstood. The good news: reducing the appearance of facial redness is possible without a prescription.
Shop Redness ReliefScience-Obsessed Skincare
You deserve skincare that makes your life better. We know that fewer ingredients means fewer flare-ups.
That’s why all our products are minimally formulated, hypoallergenic, fragrance free, dermatologist recommended, and designed with your skin in mind.
No Fragrance
No Drying Alcohols
No Steroids
No Harsh Soaps
No Parabens
No Sulfates
My quality of life improved
Before

After

Emily
Eczema Cream with Micreobalance®
My quality of life improved
"I tried lots of different lotions designed specifically for eczema. Some of them stung a little bit. Some of them even made me more itchy. Gladskin was one of the things I tried during all of this, and it was actually the only thing that gave me consistent improvement [along with removing allergens]. My skin was just so much better. My quality of life improved so much with Gladskin."
Gladskin is an absolute miracle!
Before

After

Nicole & Son Michael
Eczema Cream for Babies & Kids
Gladskin is an absolute miracle!
"My 4 month old son had severe eczema and it was heart wrenching as a mother to see him suffer and not be able to fix it. I applied Gladskin and these were the results after only one day! He is now 10 months old and eczema free!! We are life long customers."
Nicole & Son Michael
Eczema Cream for Babies & Kids
Highly recommend!
Before
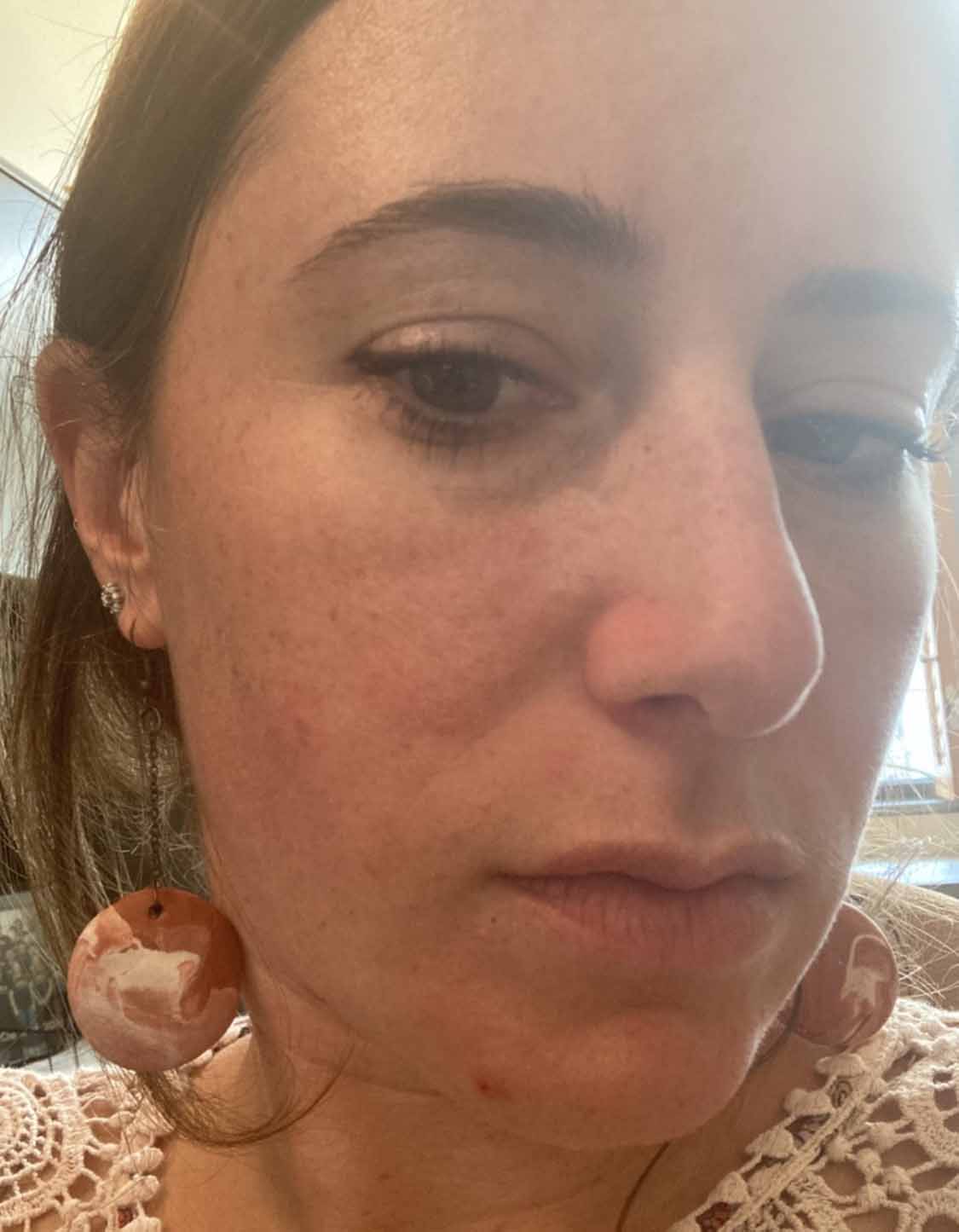
After

Stephanie B.
Redness Relief Cream with Micreobalance®
Highly recommend!
"I love this cream! My face was red and irritated, seemed to be set off by any little trigger. Within a week of using the product, I saw great results. The redness is gone. The lotion has such a calming feel on the skin."
Stephanie B.
Redness Relief Cream with Micreobalance®
As seen in

